In January of 2019 via FDA statement, Dr. Scott Gottlieb, then the Commissioner of the U.S. FDA, made a bold prediction. He foresaw that by 2025, the FDA would be approving at least ten new cell or gene therapies a year. Much as I wanted to believe this, it seemed at the time to be an extraordinarily bold projection. How does that prophecy look today, with just two years to go? In analyzing Gottlieb’s predictive power, I also want to paint a picture of a field which has reached the start of its maturation phase, progressively hitting a range of coming-of-age milestones during this period.
Talking literally of coming-of-age, I’m going to enumerate 21** commercial approvals of cell and gene therapies by the FDA. Just as one’s 21st birthday is a prestigious milestone, the medical world needs to understand that with this number of approvals, cell and gene therapy is now a grown-up field. But likewise, the field needs to behave that way as further approvals happen. And happen they will, as we see below in an analysis of the accelerating rate of approvals in recent years.
Towards the end of 2017, which I consider the beginning of the current era of cell and gene therapies, there were three new approvals. KYMRIAH®, YESCARTA®, and LUXTURNA® were indicated for relatively small but significant patient populations; they were remarkable primarily for their high level of effectiveness in previously clinically hopeless situations. Ironically, the next three years, bracketing the Gottlieb projection, constituted an approval drought, with the only two new products being ZOLGENSMA® and TECARTUS®.
For those with eyes to see, and for the rest of us in hindsight, the next two years, 2021 and 2022, saw the tide begin to turn. Breyanzi® and ABECMA®, both from Bristol Myers Squibb—a relative newcomer to cell and gene therapy—brought newfound momentum to the field. Breyanzi entered the market at least partially in competition with older CAR-T products, but with Abecma, Bristol became the first company with a CAR-T product targeting a different antigen. That alone was a significant moment for the field and gave BMS the first such cell therapy for multiple myeloma. These were followed later in the year by RETHYMIC®, which while novel, addresses an ultra-orphan population, and StrataGraft®, whose market penetration for adult patients with deep partial-thickness thermal burns remains unclear. Despite these reservations, 2021 set a record with four true cell and gene therapy approvals from the FDA.
The first approval of 2022, CARVYKTITM, from Johnson & Johnson and Legend Biotech, introduced competition for treatment of multiple myeloma by CAR-T products. Bristol now became the first company with CAR-Ts targeting distinct antigens and the first company facing direct competition for both products. Then bluebird bio, a veteran developer in the field, at last achieved not just its first, but two approvals, ZYNTEGLO® and SKYSONA®. ZYNTEGLO, in treating ß-thalassaemia, was the first hematologic product for the field and heralded true indicational broadening beyond oncology and orphan diseases. CSL’s HEMGENIX® for Hemophilia B consolidated this progress in hematology. The year ended with Ferring’s long-awaited approval of Adstiladrin®, for a total of five approvals, another record and a third consecutive year of increasing approval numbers.
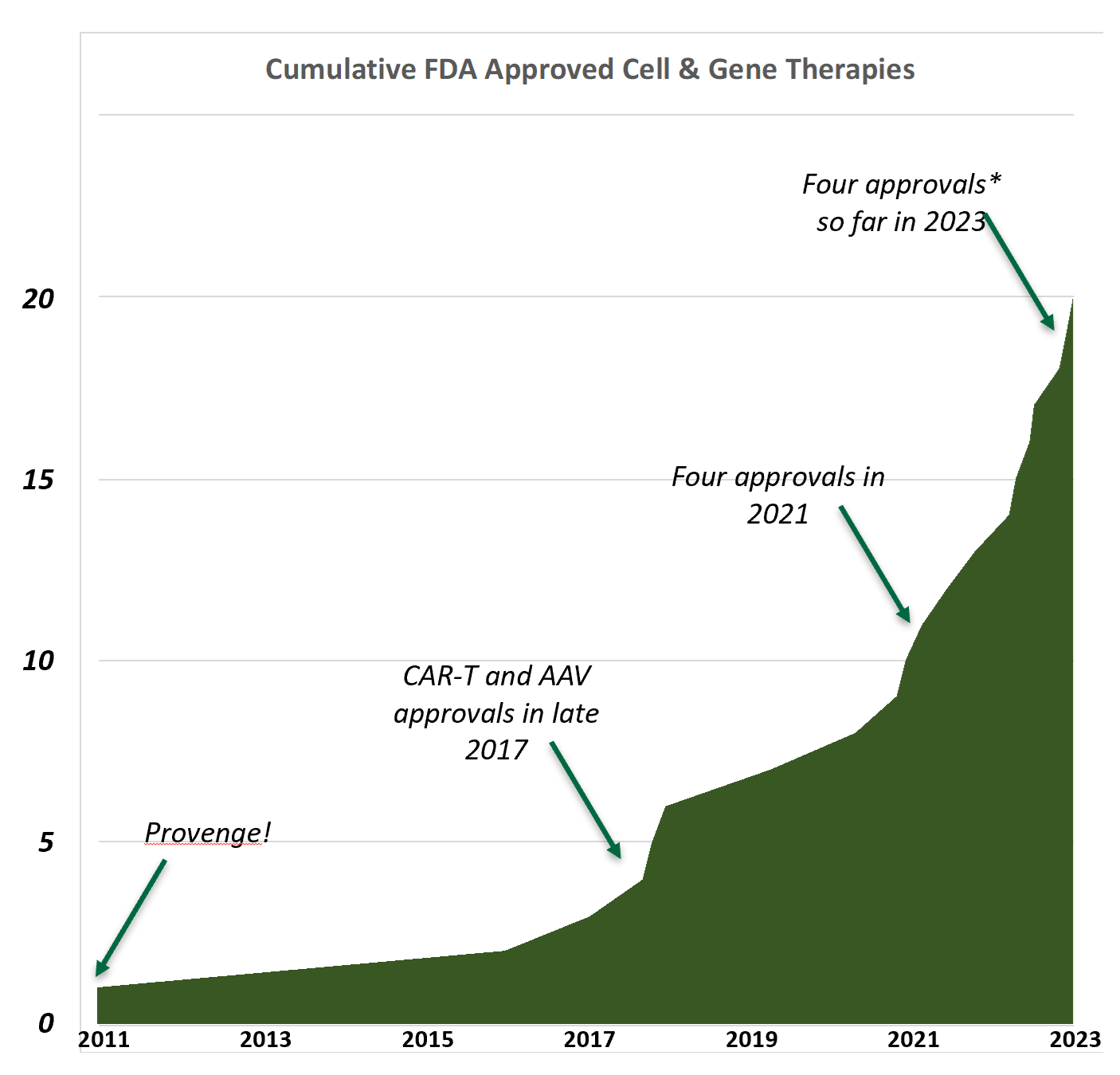
This year, we have already seen four** approvals, Omisirge®, VYJUVEKTM, ELEVIDYS and ROCTAVIANTM, and there are five publicly disclosed PDUFA dates remaining to be opined upon. These alone, if all are successful, would comprise nine commercial approvals for the year, making a fourth consecutive year of increasing numbers almost certain. It also allows us to anticipate that the field will achieve the Gottlieb projection of ten per year ahead of schedule.

21 approvals and a grown-up field: what does this mean for our future behaviour? Grown-up CMC, please: we cannot continue to advance products in this field towards commercialization without thinking through the process, the analytics, and the price. I have historically been vocal about the first two subjects, but, as we celebrate our 21st, I want to focus more attention on the last. Without closer, earlier attention this latter parameter will become cell and gene therapy’s Achilles’ heel. As connoisseurs of Greek mythology know, the Trojan War was ultimately won despite Achilles’ death, but no such outcome is guaranteed for cell and gene therapy.
There are now multiple cell and gene therapies with billion-dollar cumulative revenues (ZOLGENSMA, YESCARTA and KYMRIAH) and, by our analysis, several more heading that way soon. But too much of this revenue has been generated by the product of extremely high prices and small numbers of patients. Again, by our analysis, barely 20,000 patients have been treated with cell and gene therapies in the US, for a total revenue of $12 billion—that’s an average of $600,000 per patient, which in the long term just will not stand.
Therapeutics developers: address value and price early. Investors, plough funding into anything and everything which will reduce costs. Or, coming of age after so many hard-won battles, cell and gene therapy will need more than a horse, Trojan or Dark, to win the war.

* Acknowledging countless technical discussions with Senior Consultant Christina Fuentes, Ph.D. and insight into revenues and market penetration from Principal Benjamin Nelson and Consultant Madeline St. Onge, MBA.
** We have excluded multiple products from this count which have been technically approved as cell therapy products by the FDA. The two currently off-market products Laviv and Gintuit are excluded because they are not currently available to patients. We also exclude the eight cord blood-derived products from Cleveland Cord Blood Center, Duke University School of Medicine, New York Blood Center, Clinimmune Labs, University of Colorado Cord Blood Bank, MD Anderson Cord Blood Bank, LifeSouth Community Blood Centers, Inc. and Bloodworks, as well as the recently approved Lantidra. For reasons too complex to detail here, related to the small extent of their manufacturing manipulation and their homologous use, we do not consider these products to be ‘true’ cell therapies. Indeed, we further question whether they should even be regulated by CBER, but rather as transplants under NOTA-type legislation, but that is a subject for another day.
Subscribe to our mailing list for the latest insights on advanced therapy development, regulatory updates, industry trends, and upcoming events from Dark Horse Consulting Group.
We respect your privacy. Unsubscribe at any time. We will never sell your information.




